QHerit® carrier screening
Broad portfolio of clinically relevant, right-sized solutions for patients
As patients consider starting a family, they may want to have a better understanding of their genetic makeup, and whether they are at increased risk of passing along a genetic variant to their children. Genetic carrier screening is a crucial tool to help couples have a better understanding of risk and guide their family planning decisions.
Relevant results to help you provide
confident carea
As part of our commitment to providing clinically relevant, right-sized solutions, our QHerit® product portfolio has been designed to help you understand your patient’s genetic risks and support family planning discussions.
Our QHerit panels are ever-evolving, and we currently offer 5 screening panel options for up to 611 diseases to help you to select a medically appropriate panel based on your patient’s needs.


Pan-ethnic carrier screening with QHerit
Our panels are designed with American College of Obstetricians and Gynecologists (ACOG) guidelines and American College of Medical Genetics and Genomics (ACMG) Practice Resources in mind.
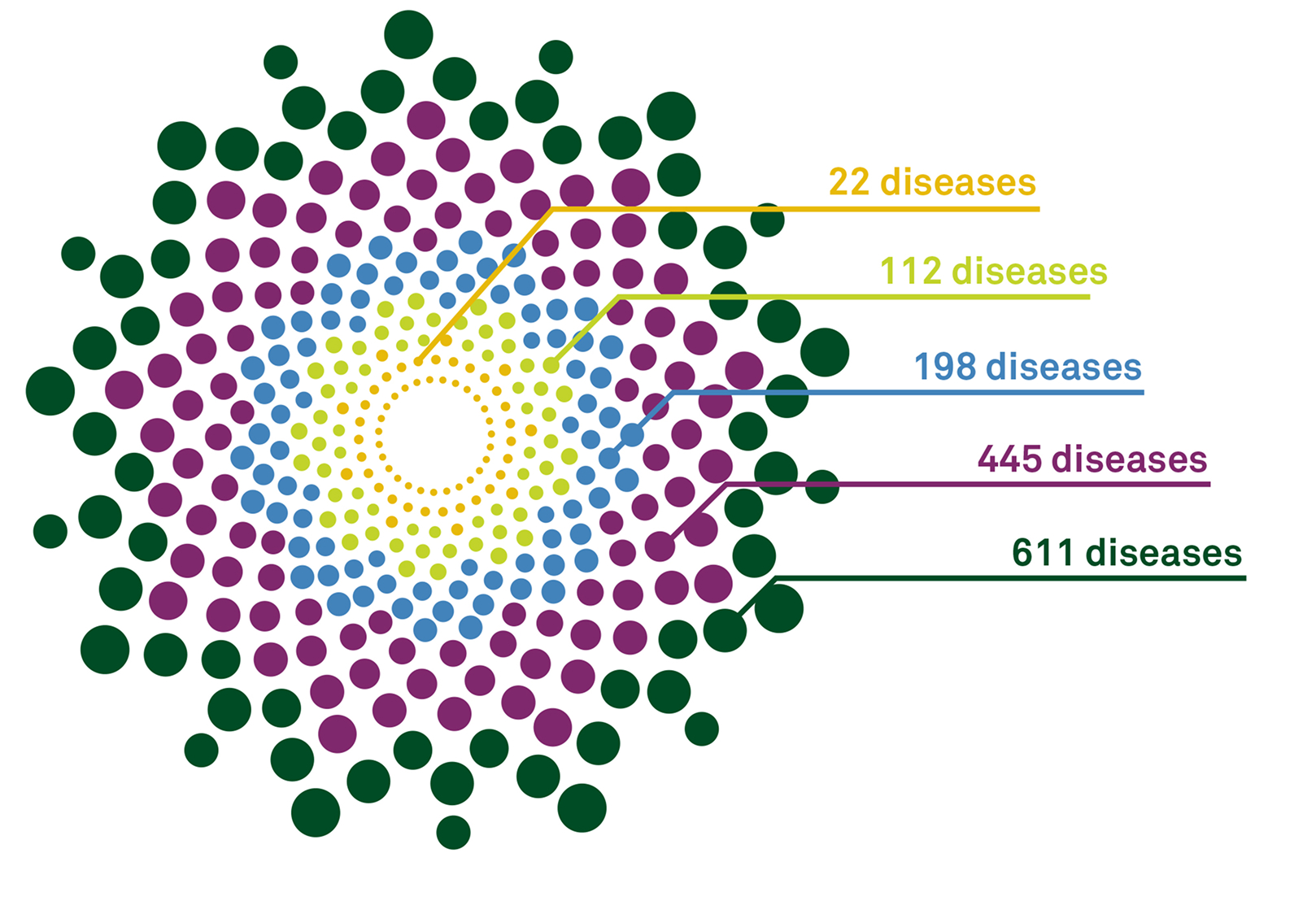
22 diseasesc
24 genes
112 diseasesc
113 genes
198 diseasesc
199 genes
445 diseasesc
446 genes
611 diseasesc
612 genes
The most common diseases including cystic fibrosis, spinal muscular atrophy, fragile X, and Tay-Sachs
Recommended genes listed by ACMG tier 3, which includes the 24 genes in the smaller panel
Builds on the 113-gene panel with 86 additional genes
An additional 247 genes included plus the 199-gene panel
Our most comprehensive panel with 166 additional genes offering the greatest insights
Female: 14232
Male: 14227
Female: 14230
Male: 14231
Female: 13832
Male: 13831
Female: 14228
Male: 14229
Individual component tests of the QHerit panels are available to be ordered separately. Download the PDF to see the complete list.
22 diseasesc
24 genes
112 diseasesc
113 genes
Male: 14227
198 diseasesc
199 genes
Male: 14231
445 diseasesc
446 genes
Male: 13831
611 diseasesc
612 genes
Male: 14229
Individual component tests of the QHerit panels are available to be ordered separately. Download the PDF to see the complete list.
View all the diseases and genes included in QHerit
Download PDF
Empowering you and your patients to optimize family planning decisions
We understand that you and your patients depend on these results for family planning and care. QHerit provides medically appropriate and clinically relevant insights that you and your patients need to help plan their futures.


Genetic testing that’s accessible and affordable
We believe cost shouldn’t be a barrier to care. We are committed to keeping costs low so that more patients, regardless of their financial status, can access the screening they need.
- Access to ~90% of insured lives nationwide and preferred lab network status with major health plans including Aetna®, Horizon®, and UnitedHealthcare®
- No surprises with a bill, as we’ll contact you and/or your patient if the cost is expected to exceed $299
- Dedicated team of Patient Navigators works directly with patients to review insurance coverage, answer billing questions, and clarify costs for QHerit carrier screening
- QHerit Supplemental Financial Assistance Program is available for both insured and uninsured patients who qualify

Helping her live her healthiest life
Visit the Test Directory to review the QHerit carrier screening panel options for your patients
a QHerit® 22, QHerit 112, QHerit 198, QHerit 445, and QHerit 611 are carrier screening tests, and they screen for variations in genes linked to certain health disorders that can be passed from parents to children. QHerit 22 screens for 24 genes, QHerit 112 screens for 113 genes, QHerit 198 screens for 199 genes, QHerit 445 screens for 446 genes, and QHerit 611 screens for 612 genes. For a full list of genes that each panel in the QHerit family screens, visit QHerit.com. If the results from any panel in the QHerit family suggest that a patient may be a carrier of a gene variation that can cause a health disorder in her offspring, it is recommended that her reproductive partner be offered genetic screening, and that genetic counseling be provided. Pregnancy management decisions should not be based on the results of these screening tests alone. As with any test, there may be false positives or false negatives. The positive predictive value of the screening test varies by genetic variation, and may be lower for rare conditions. Each panel in the QHerit family is a laboratory-developed test that has been developed and validated pursuant to the Clinical Laboratory Improvements Amendments of 1988 (CLIA) and, as such, it has not been reviewed by FDA.
b QHerit panels are screening tests. QHerit does not diagnose a disease or disorder.
c Panel components for males do not include specified X-linked diseases.
d Based on Quest Diagnostics full year 2023 claims analysis.
Test codes may vary by location. Please contact your local laboratory for more information.
While we offer a comprehensive testing menu, some patients may have an interest in screening for a specific disorder, such as cystic fibrosis. For these patients, Quest Diagnostics offers single-gene screening. Consultation available on genetic test selection and results interpretation 1.866.GENE.INFO (1.866.436.3463).
Please note that Quest offers a variety of single-gene and gene panel testing. For the genetic panels noted in this webpage, there may be single-gene tests or smaller panels that may be applicable for your patient. Refer to the Quest Diagnostics Test Directory for further information.



Genetic consultation available
Genomic science specialists are available to help with test selection and results interpretation 1.866.GENE.INFO (1.866.436.3463)
Patient info
For assistance with questions about the testing process or locations, call 1.866.MYQUEST (1.866.697.8378)
Call 1.866.MYQUEST
(1.866.697.8378)
Thank you for your submission
The brochure will be downloaded automatically.